



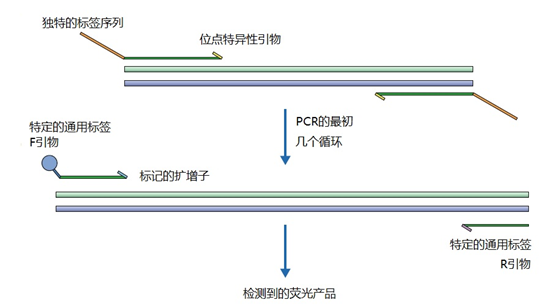
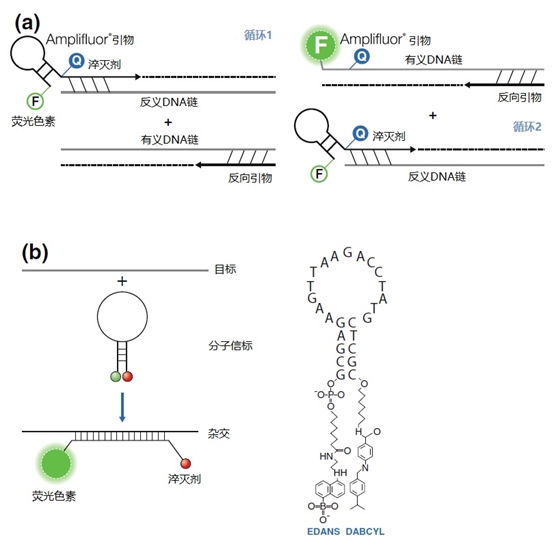
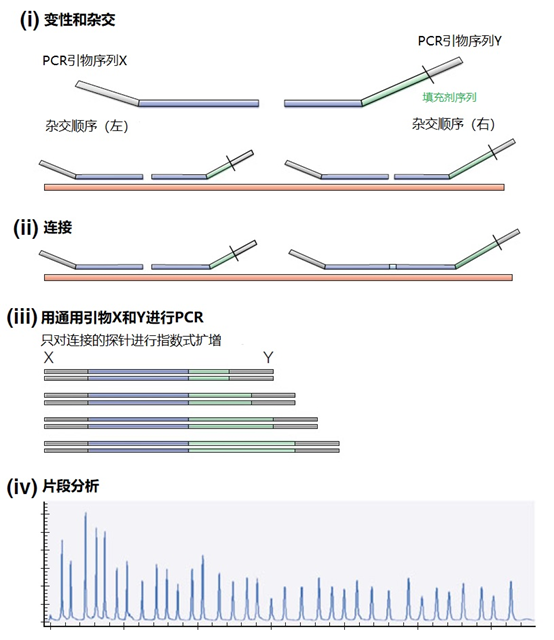
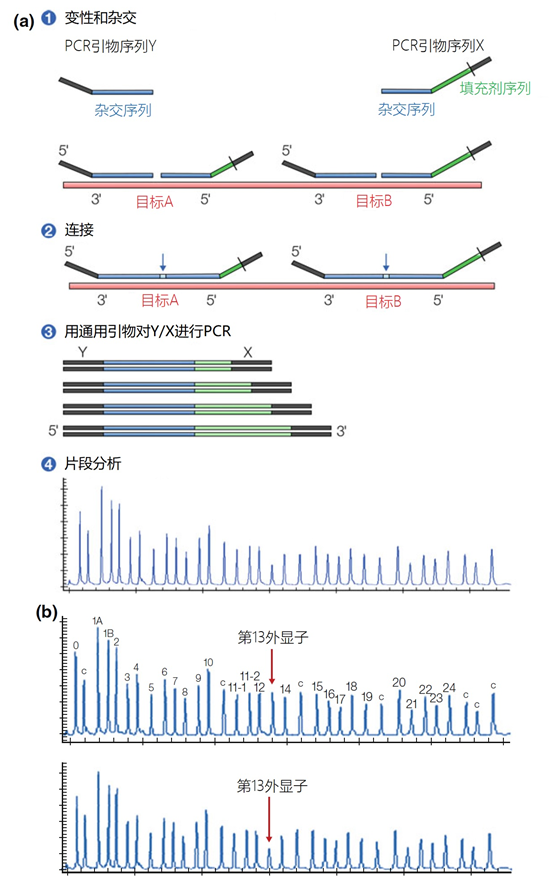
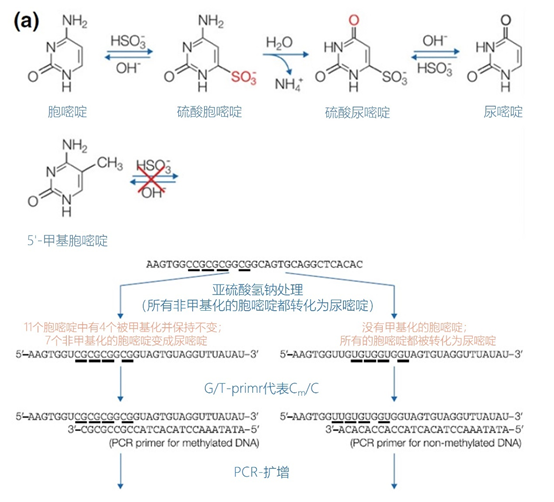
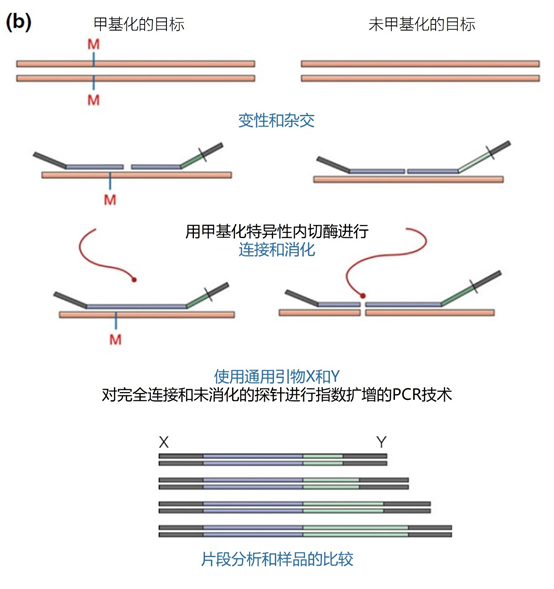
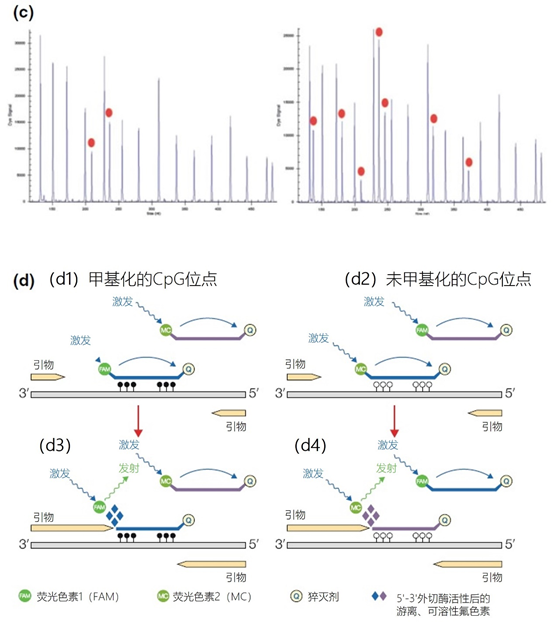
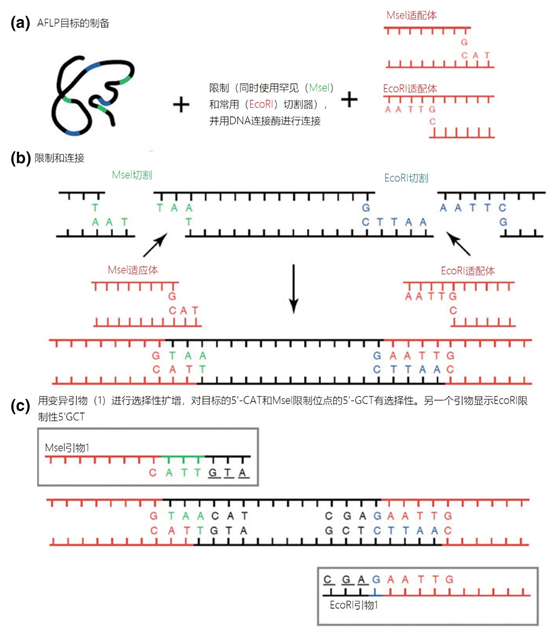
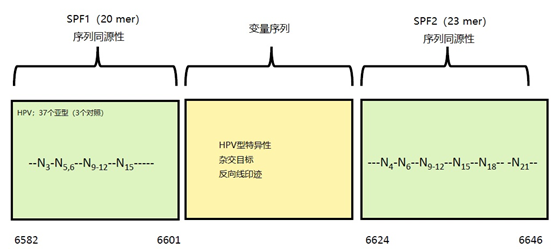
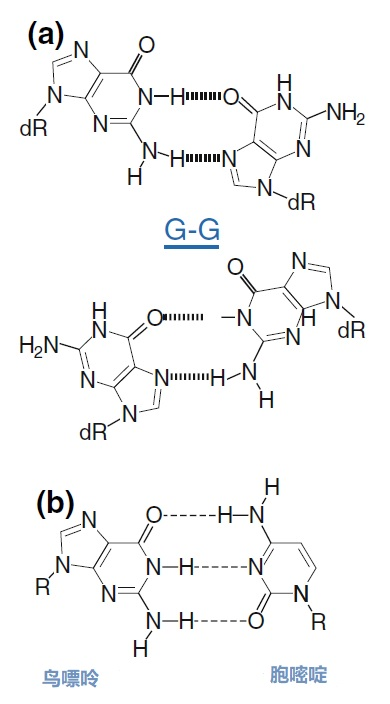
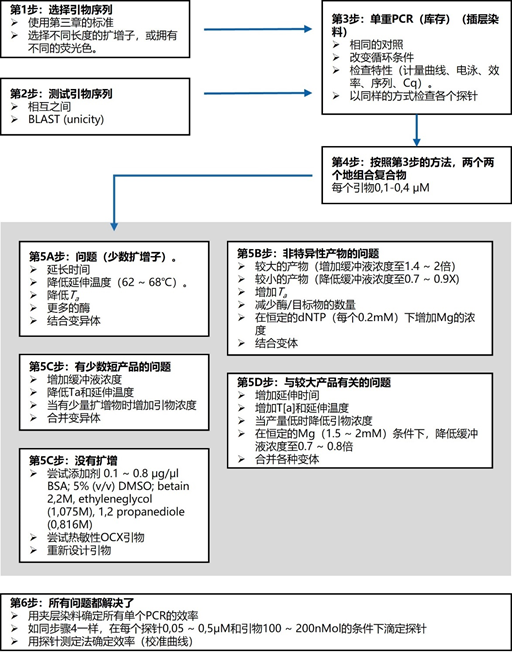
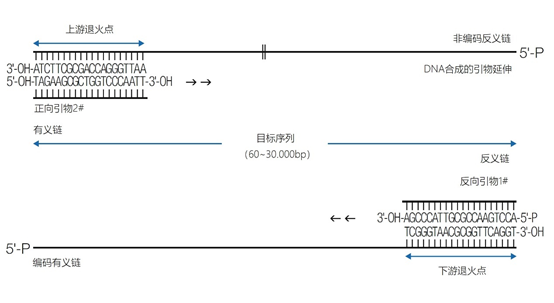
主要而言,引物的大小约为20个核苷酸,与靶点完全互补,经常用于PCR。然而,确实存在很多为特殊应用设计的引物变异。例如,这些引物变异可以更长,可以拥有特殊的功能、错配、标签或碱基替代物(图1)。它们可以为识别特殊的突变(如易位)而设计,也可以满足特殊的标准(如在多重配置中没有交叉杂交)。 图1 | 不同类型的引物 a 最小引物:引物,与靶点不完全匹配(也叫变性引物)。错配并不总是像预期的那样影响杂交体的稳定性。在某些错配中存在对稳定性的一些贡献。 b 外加引物:一个带有5′外挂的引物,以便将额外的序列附加到扩增物上。该序列不与本地靶点退火。这个延伸序列通常用一个间隔物与5′末端的序列相连。这个延伸序列也将在随后的循环中被扩增(见图2)。一个额外的功能被添加到扩增物中,如用于克隆的限制性位点或用于蛋白质合成的ATG起始密码子,扩增物作为一个基质发挥作用。 1、扩展引物 扩展引物在5'端含有额外的核苷酸,不与靶点杂交,在第一个PCR循环中不被复制。这些引物在第二个PCR循环后成为扩增物的一个组成部分。通过这种方式,可以添加额外的序列,如限制性位点、翻译起始序列或启动子序列,最多为50个碱基。 一种特殊的扩展引物类型配备了一个编码标签,其序列与问题相适应。这个标签可以作为标签互补引物或探针的杂交靶点(图2)。 图2 | 带有5′-外挂的标签杂交原理 在基因座特异性引物对上添加一个5′标签。该标签由基因组上不存在的序列组成。当标签成为扩增物的一部分时,就形成了一个新的靶点序列。一个带标签的特异性引物将接管扩增反应。标签有助于在与正向引物耦合的荧光色素的基础上进行识别。MLPA(见图4)使用标签扩增的原理。 标签也可以被设计成显示出独特的能力,例如形成发夹环。发夹引物利用这种构造来猝灭非退火引物的信号。扩增后,标签将最终成为扩增物中的一个线性序列。这样一来,猝灭剂和荧光剂就会分离,从而导致荧光的产生(图3a)。同样的原理适用于猝灭的探针(图3b)。有些标签有独特的序列,在自然界中不存在。通过使用多个标签,可以同时捕获多个扩增物。 图3 | 实时PCR中引物和探针的猝灭 引物(a)和探针,即分子信标(b)在溶液中不发亮,这是猝灭的结果。报告基团和猝灭者在彼此附近,从而交换激发电子(猝灭)。一旦发夹引物在PCR过程中被拉长(a)或探针与扩增物退火(b),荧光剂和报告基团将被物理分离并发生荧光。 多重连接依赖性探针扩增(MLPA)可使用另一种类型的扩展引物(图4)。 图4 | 多重连接依赖性探针扩增(MLPA) 该技术中使用了标签(见图2)。反应从双重杂交开始(探针的蓝色部分)。两个探针以头对尾的构象同时与靶点杂交。连接酶连接两个探针的 3'-5'-末端。标签序列 X 和 Y 以及填充序列(S)不杂交。每个探针组具有不同的唯一填充序列长度(S1、S2、S3 等)。这种独特的长度便于识别每个放大器。所有连接的探针都用靶向序列 X 和 Y 的相同引物对进行扩增。通过毛细管电泳区分扩增子的填充长度。 MLPA是一种PCR技术,两个探针在靶点上头尾相接地杂交。独特的标签位于两个探针的3ʹ(右)或5ʹ(左)侧。两个标签都包含一个通用引物序列(X和Y)。此外,在5'侧有一个特殊的标签,即所谓的填充序列。 这个缓冲序列的长度是可变的。引物和填充物的序列没有相互作用,如果需要,可以使用几十个不同长度的序列。在靶点杂交后,相邻的探针被连接起来。现在,可以使用与X和Y互补的引物与悬吊的标签退火,进行PCR。合成的扩增物(对10-30个不同靶点的特异性)可以通过(毛细管)电泳和/或浓缩来分离(见图5和图6c)。 图5 | 多重连接依赖探针扩增法(MLPA) a 用于检测靶点序列A和B的MLPA法原理。两个完全匹配的(半)探针与靶点A或靶点B杂交,每个探针都有一个带有3′引物退火位点的标签(上游)。下游不仅有一个引物退火位点,而且还有一个所谓的填充序列,其长度可以变化。杂交后,探针通过一个5′-3′连接酶的磷酸糖桥进行共价连接。这样就产生了一个新的靶点序列,由引物退火位点、独特序列和填充序列组成。在所有连接的探针变性后,开始进行PCR。由于两个半探针的连接只发生在3′-5′的完美匹配处,PCR产物也只有在这时产生,这决定了这种方法的高选择性。不同填料的长度差异使得产品可以按大小进行分离。 b 毛细管电泳对PCR产物的分辨率足以检测同源性或杂合性,或者可以检测到完全缺失。第二行是BRCA1基因外显子13缺失的患者与正常质控组的对比。 图6 | 甲基化特异性PCR:原理和三种不同的检测方法 三种方法中的每一种都需要对胞嘧啶进行亚硫酸氢盐处理并将C转化为U。这样可以区分甲基化的CpG位点(保留C)和非甲基化的胞嘧啶(变为U)。 b MS-MLPA(甲基化特异性MLPA),可以检查40个位置的甲基化状态。在亚硫酸氢盐处理后的两个反应中使用两个探针。左边是一个带G的半探针的例子,可以检测到C没有变化的甲基化靶点,右边是一个带A的半探针的例子,可以识别非甲基化的位置。就像图5中一样,填料序列的长度不同。在毛细管电泳后,可以确定(副产品的长度)是否涉及同型、异型或野生型样品。 c 传统的PCR,有两种不同的测定。在「C」在亚硫酸氢盐处理后未被甲基化的位置,「C」在亚硫酸氢盐处理后未被甲基化。 在亚硫酸氢盐处理后「C」未被甲基化的位置,「C」已被转化为「U」。引物,一种是带有G/A取代的碱基的测定,另一种是带有原始「G」的引物,将选择性地结合。如果有甲基化的部分,用含有G的引物进行PCR。当C被转化为U时,用另一个引物(用A代替G)进行PCR。PCR本身是用普通的dNTPs完成的。 也可以应用一个能与新引物杂交的序列;如扩增片段长度多态性(AFLP)技术(见图7)。 图7 | 扩增片段长度多态性(AFLP) AFLP是通过扩增不同的限制性片段来获得全基因组的图像,随后通过毛细管电泳进行分析。片段的模式可以是物种、类型或亚类型特定的 首先,用限制性酶处理基因组,造成两边都有粘性末端的双链片段。使用常用切割器(作用于相对常见的序列,如EcoRI)和罕见切割器(相对罕见的序列,如MseI,TaqI)的组合。这两种酶之间的比例决定了产生的片段数量 (a) 互补适应体与「粘性末端」杂交。使用连接酶(b)将适配体和限制性位点连接到基因组DNA上。通过这种方式,DNA被扩展成可以结合互补引物的通用序列(c)。此外,引物可以用1-3个额外的碱基进行扩展,限制了PCR产物的数量。在c中,引物被提供了三个额外的碱基(在此图中「GTA」代表MseI-引物,「AGC」代表EcoRI-引物)。完全(100%)的互补性将只出现在3′侧有限的地方;只有这样,引物才会被扩展为扩增物。 2、退化/共识引物 退化引物或共识引物与靶点序列不完全互补。这些引物缺少一个或多个核苷酸或在某些位置上有一个或多个核苷酸的差异。尽管有一定数量的错配,这些引物被设计成退火。由于这种不完全的碱基配对(较少的H桥,较少的堆叠),亲和力下降(图8)。 图8 | 37种HPV类型和3种质控的简化核苷酸序列比对 从病毒性HPV基因组的6582至6646位进行HPV类型的核苷酸序列比对 正向(SPF1)和反向(SPF2)用绿色矩形表示。37种HPV类型之间发现不同碱基的位置用「N」表示,排列中的同源性用「-」表示。大多数序列有3-5个不明确的地方。正向和反向序列位置的3′侧具有大多数同源性。针对HPV16的引物SPF1/2将导致完全匹配。引物中6592和6595位置的肌苷的吸收避免了与其他类型的不匹配。引物浓度和PCR条件的选择是为了使引物能与其他类型的HPV进行交叉杂交。 位置6601和6624之间的区域(黄色矩形)是高度可变的。它包含类型特异性探针的热点,可以区分使用SPF1/2引物对合成的所有37种HPV类型的扩增子。(关于碱基组成,见图2) 由于3'端的核苷酸差异阻碍了PCR的进行,引物的设计需要将5'端的非互补段与3'端的完全匹配放在一起。也可以设计一个引物,在有碱基差异的位置上有一个随机碱基或SNP的特殊组合特征。例如,「R」(A或G),「B」(C或G或T)或「N」(任何碱基);正确的字母可在IUPAC/FASTA代码中找到。 退化的引物通常较长,以补偿杂交能力的损失。显然,错配不是绝对的;G-G、T-T、A-A和C-C之间的任何H键都有可能。然而,这将导致螺旋结构的变形和不太稳定的杂交体(图9)。 特别是在微生物研究中,以及在淋巴瘤/白血病的诊断中,可能需要用一对引物检测多个靶点(例如,使用16S rRNA基因上的类似序列的若干变异基因型的病毒或多个细菌物种)。这种引物被称为共识引物;但实际上,这些引物也被称为退化引物。 可以用肌苷替换引物中某些关键位置上的错配核苷酸。这个碱基自然存在于tRNA中,并与A、U和C化学上「匹配」。因此,肌苷可以与某些错配杂交,但对特异性没有任何贡献。 3、转位引物 转位引物的设计是为了在转位部位的上下游进行杂交。最为人所知的是恶性肿瘤的易位,其中有些肿瘤具有特定的亚型,适合于分子诊断。 在易位时,不同的染色体重新组合成一个完整的新序列。易位引物会在断裂点的两边进行杂交,从而确保只发生与易位有关的扩增。它们不会扩增没有易位的野生型序列。 4、多重PCR 在多重PCR策略中,可以在一个PCR反应中同时检测多个靶点。多重PCR使用2对以上的引物,在一个PCR反应中实施5对甚至10对引物,可以有效(或适当)使用试剂和仪器;相对降低了成本,减少了工作量。用于多重PCR的引物符合对普通引物有效的相同规格。 此外,所有可能的PCR反应将在相同的条件下进行,相当于反应间的差异。此外,与其他每个引物形成双链的风险必须是最小的。要求与靶点有同样强的结合力(相同的Tm),也要求每个单独的单倍体PCR反应有相当的效率。由于某些引物在与其他引物形成双倍体后可能出现假阴性,导致无法与靶点退火,因此避免这种双倍体的形成至关重要。关于这最后一个标准,引物通常要长一些,需要非常仔细地选择所需的引物浓度(见图10)。 图10 | 优化多重设计的阶段和可能性。传统的PCR需要PCR后的步骤,如电泳和杂交。实时PCR方案的开发始于使用插层染料,如SYBR® Green,并结合熔解曲线分析。反应效率将使用参考曲线确定。下一阶段是对探针的优化。 多重方法在微生物学中经常被用来从活检、拭子、尿液、粪便或其他样品中的许多可能的病原体中挑选出一个。另一个应用是血液学恶性肿瘤的基因定量。 对于所有的多重PCR方案,强烈建议首先在干试验中测试所有的引物,以估计引物在单倍体PCR反应中的表现以及随后在多重PCR格式中的表现。此外,还必须在实践中使用适当的质控样品测试所有引物在多重PCR方案中的正确活性。 5、重复引物 在大多数诊断PCR反应中,一个引物对用于扩增一个独特的靶点序列。少数诊断引物被设计成在基因组上的多个位置与相同的靶点杂交,并在同一试管中为一些不同的PCR反应提供引物。穿插重复序列,如广泛存在的简单序列重复(SSR)包含长段的简单序列。 一个著名的小型SSR的例子是(CACA)n-repeat和它的引物与重复的(GTGT)m。由于存在许多重复,引物在所有这些位点退火,初始化DNA合成将在大约40个PCR循环后产生不同大小的扩增物池(见图11)。 图11 | PCR中的引物退火点、合成方向和靶点序列 该图中描述了最重要的术语。传统PCR的靶点通常为60-400个碱基;而实时PCR的靶点为80-150个碱基。 这种「重复间」PCR中扩增物的电泳模式,即所谓的指纹,是基因组特异性的,可用于基因分型。 同样的原则也可适用于大中型重复序列(10-300 bp),如ERIC重复序列和各种BOX序列,这些序列经常用于微生物学分型。 来源: 诊断科学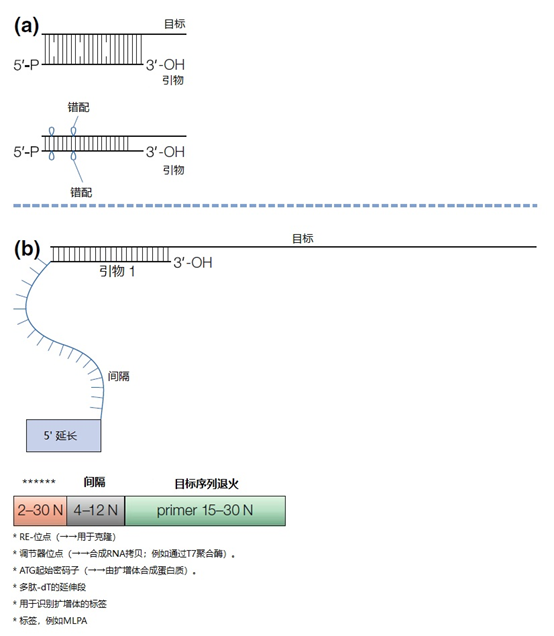
声明:本平台注明来源的稿件均为转载,仅用于分享,不代表平台立场,如涉及版权等问题,请尽快联系我们,我们第一时间更正,谢谢!


