



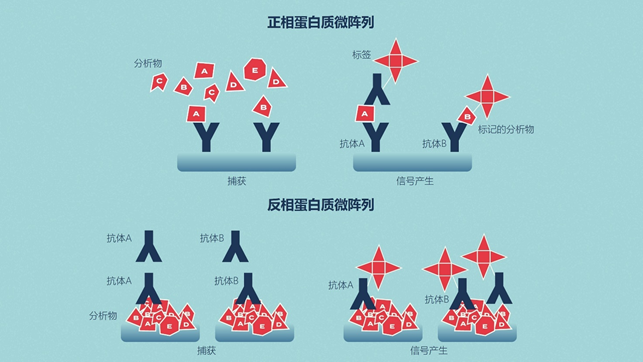
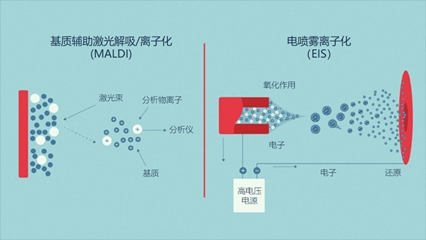
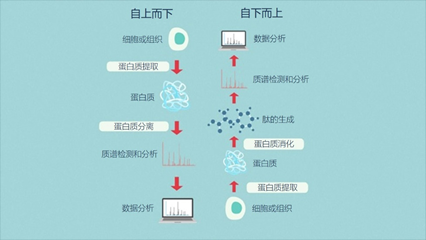
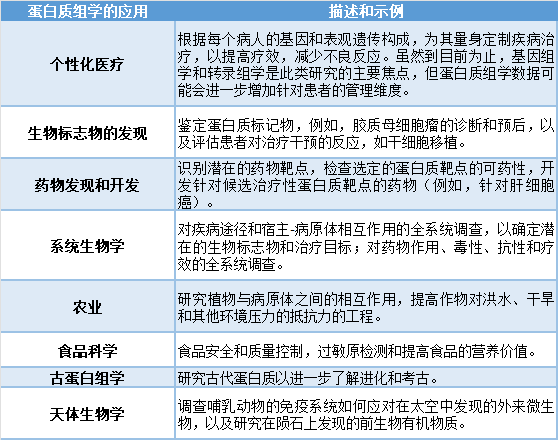
01 什么是蛋白质组? 蛋白质是由称为氨基酸的构建块组成的生物分子。蛋白质是生命所必需的,具有结构、代谢、运输、免疫、信号和调节等许多作用[1]。 术语“蛋白质组”是由澳大利亚博士生Marc Wilkins于1994年在意大利锡耶纳举行的研讨会上提出的[2]。它是一个概括性的术语,指的是一个生物体可以表达的所有蛋白质。每个物种都有自己的、独特的蛋白质组。 与基因组(每个生物体内的全套基因)不同,蛋白质组的组成随着时间和整个生物体的变化而不断变化[3]。因此,当科学家提到蛋白质组时,他们有时也是指某一特定时间点的蛋白质组(如胚胎与成熟生物体),或指生物体内某一特定细胞类型或组织的蛋白质组。 图1 | 人类基因组中大约有2万个基因,人类转录组中大约有10万个转录物,人类蛋白质组中超过100万个蛋白质形态 02 什么是蛋白质组学? 蛋白质组学是对蛋白质组的研究,研究不同的蛋白质之间如何相互作用以及它们在生物体内发挥的作用[4]。 虽然蛋白质的表达可以通过研究mRNA的表达来推断,mRNA是基因和蛋白质之间的中间人,但mRNA的表达水平并不总是与蛋白质的表达水平有很好的相关性[1,3]。此外,对mRNA的研究并不考虑蛋白质的翻译后修饰、裂解、复合物的形成和定位,或可以产生的许多变体mRNA转录本;所有这些都是蛋白质功能的关键。 1975年,随着二维蛋白电泳技术的发展,第一批符合“蛋白质组学”研究标签的实验开始进行[5]。 然而,只有在20多年后,随着质谱技术的发展,才有可能对每个样品的多个蛋白质进行真正的高通量鉴定[6]。 从那时起,质谱的灵敏度和准确性已经发展到可以可靠地检测到低至阿托摩尔范围的蛋白质(每10^18个分子中有1个目标蛋白分子)[7],而且其他各种蛋白质组学技术也得到了发展和优化。 03 蛋白质组学可以回答哪些关键问题? 广义上讲,蛋白质组学研究在蛋白质水平上为健康和疾病的细胞过程提供了一个全局的视角[3,4]。为此,每项蛋白质组学研究通常都会在目标生物体的蛋白质组中一次集中研究以下一个或多个方面,以慢慢建立现有的知识: 04 蛋白质组学技术 4.1、低通量方法 4.1.1、基于抗体的方法 诸如ELISA(酶联免疫吸附试验)和Western blotting等技术依赖于针对特定蛋白质或表位的抗体的可用性,以识别蛋白质并量化其表达水平。 4.1.2、基于凝胶的方法 二维凝胶电泳(2DE或2D-PAGE)是最早开发的蛋白质组学技术,它使用电流根据蛋白质的电荷(第一维)和质量(第二维)在凝胶中分离。差分凝胶电泳(DIGE)是2DE的一种改良形式,使用不同的荧光染料,可以在同一凝胶上同时比较两到三个蛋白质样品。这些基于凝胶的方法被用来在进一步分析前分离蛋白质,例如质谱分析(MS),以及用于相对表达谱分析。 4.1.3、基于色谱的方法 基于色谱的方法可用于从复杂的生物混合物(如细胞裂解液)中分离和纯化蛋白质。例如,离子交换色谱法根据电荷分离蛋白质,尺寸排除色谱法根据分子大小分离蛋白质,亲和色谱法采用特定的亲和配体和其目标蛋白质之间的可逆相互作用(例如,使用凝集素纯化IgM和IgA分子)。这些方法可用于纯化和鉴定感兴趣的蛋白质,以及为进一步分析准备蛋白质,如下游的MS[8]。 4.2、高通量方法 4.2.1、分析微阵列、功能微阵列和反相微阵列 蛋白质微阵列将少量的样品应用于“芯片”进行分析(有时是以玻璃片的形式,表面经过化学修饰)。 特定的抗体可以被固定在芯片表面,用于捕获复杂样品中的目标蛋白。这被称为分析蛋白微阵列,这些类型的微阵列被用来测量样品中蛋白质的表达水平和结合亲和力。 功能蛋白微阵列被用来描述蛋白功能,如蛋白-RNA相互作用和酶-底物周转。在反相蛋白质芯片中,来自例如健康与病变组织或未处理与处理过的细胞的蛋白质被结合到芯片上,然后用针对目标蛋白质的抗体对芯片进行检测。 图2 | 正相和反相蛋白质芯片的区别 4.2.2、基于质谱的蛋白质组学 有几种“无凝胶”的方法来分离蛋白质,包括同位素编码亲和标签(ICAT)、细胞培养中的氨基酸稳定同位素标记(SILAC)和相对和绝对定量的同位素标签(iTRAQ)。这些方法既可以进行定量,也可以进行比较/鉴别蛋白质组学。 还有其他一些定量较少的技术,如多维蛋白质鉴定技术(MudPIT),其优点是更快和更简单。其他无凝胶的、用于蛋白质分离的色谱技术包括气相色谱法(GC)和液相色谱法(LC)[8,9]。 4.3、质谱分析工作流程 无论蛋白质样品是如何分离的,下游的质谱工作流程包括三个主要步骤: 1. 蛋白质/肽被质谱仪的离子源离子化; 2. 产生的离子根据其质量和电荷比被质量分析器分离; 3. 离子被检测。 当使用MS上游的无凝胶技术,如iTRAQ或SILAC,样品直接用于输入质谱仪。当使用基于凝胶的技术时,蛋白质点首先从凝胶中切出并被消化,然后用LC分离或直接用MS分析。 有两种主要的电离源,即: ➤ 基质辅助激光解吸/离子化(MALDI); ➤ 电喷雾离子化(ESI)。 图3 | 基于MS的蛋白质组学的两个主要电离源 其他不太常见的来源包括化学电离、电子冲击和辉光放电电离。 有四种主要的质量分析器: ➤ 飞行时间(TOF); ➤ 离子阱; ➤ 四极杆傅立叶变换离子回旋加速器(FTIC); ➤ 扇形电场和扇形磁场是另外两种不太常用的质量分析器。 05 什么是串联质谱? 肽可以进行多轮破碎和质量分析,这一过程被称为串联MS、MS/MS或MSn。通过将相同或不同的质量分析器串联起来,如四极杆-TOF(Q-TOF)或三极杆(QQQ)MS,可以利用不同质量分析器的优势,进一步提高整个蛋白质组分析的能力。 简单的质谱设置,如MALDI-TOF仅用于肽的质量测量,而串联质谱仪则用于确定肽的序列。 06 自上而下蛋白质组学 与自下而上蛋白质组学 在自上而下蛋白质组学中,感兴趣的样品中的蛋白质首先被分离,然后再被单独表征[1,10]。 在自下而上蛋白质组学中,也被称为“鸟枪法”蛋白质组学,样品中的所有蛋白质首先被消化成复杂的多肽混合物,然后对这些多肽进行分析以确定样品中存在哪些蛋白质[1,10]。 自上而下和自下而上的方法都有各自的优点和缺点,以及各自更适合的应用[10,11]。例如,自上而下的质谱更适合于研究不同的PTMs和蛋白质的异构体。然而,它受到分离复杂的蛋白质混合物所固有的困难和MS对较大的蛋白质(特别是 > 50到70kDa)的敏感性下降的限制[1]。 相比之下,虽然自下而上的质谱中使用的肽(长度约为5到20个氨基酸)更容易分馏、离子化和片段化,但这种方法提供了对样品中最初存在的蛋白质的间接测量,并且严重依赖推断[1]。 一种混合的“自下而上”的方法已经被开发出来,它采用了比传统的自下而上蛋白质组学更大的肽段,从而有可能允许更多独特的肽段匹配。 图4 | 自上而下和自下而上蛋白质组学之间的差异。 07 蛋白质组学的数据分析 蛋白质组学研究,特别是采用高通量技术的研究,可以产生大量的数据[12]。除了产生的大量数据外,蛋白质组学的数据分析对于某些技术来说也是相对复杂的,如鸟枪法质谱[13]。更加复杂的是可用于蛋白质组分析的生物信息学工具的范围[14-17]。 蛋白质组研究人员在试图优化他们的蛋白质组数据的存储和分析方式时,面临着许多障碍[12]。 在计划蛋白质组实验时,科学家不仅需要考虑试剂和实验室设备的成本,还需要考虑数据存储和分析的成本,他们还必须评估所需的生物信息学技能和计算资源水平。 蛋白质组学研究往往需要多个数据处理和分析步骤,需要按照特定的顺序进行[12]。为了满足这一需求,研究人员越来越多地将所需的脚本、工具和软件组装成适合其特定研究问题的定制蛋白质组分析管道。 08 蛋白质组学的应用 蛋白质组学的应用多得令人难以置信,而且种类繁多。下表列出了其中一些应用: 09 蛋白质组学的未来 目前,蛋白质组学工作流程在很大程度上依赖于质谱[1]。尽管这项技术已被证明是强大的,但研究人员现在正在展望蛋白质组学的未来,即“超越质谱”。尽管质谱的灵敏度很高,但样品中仍需要有数百万的目标分子才能被检测到。这意味着低浓度的目标分子(如血清生物标志物)在复杂的环境中(如人类血清)可能无法检测到,除非首先富集。 科学家们仍在寻找高通量蛋白质组技术的解决方案: 1) 在目标蛋白质组的动态范围内具有出色的灵敏度(例如,人类蛋白质组的灵敏度为10^7); 2) 可以直接读取整个蛋白质序列并识别其PTMs,以及; 3) 不需要从理论蛋白质匹配数据库中进行推断[1]。 有几种有前途的技术,虽然目前受到灵敏度、通量或成本的限制,但可能会在蛋白质组学领域占据主导地位[1]。这些技术包括新生的荧光指纹方法和尚未开发的用于蛋白质高通量单分子测序的亚纳米孔阵列。 随着蛋白质组学技术的发展,蛋白质组学数据分析的方法也将同样快速发展。例如,云计算、软件容器和工作流系统等数据技术的发展势头强劲,这些技术将使人们能够自由的获得用于蛋白质组数据分析的顶级计算资源,而不受研究人员的位置、IT基础设施或计算专长的影响[12,18,19]。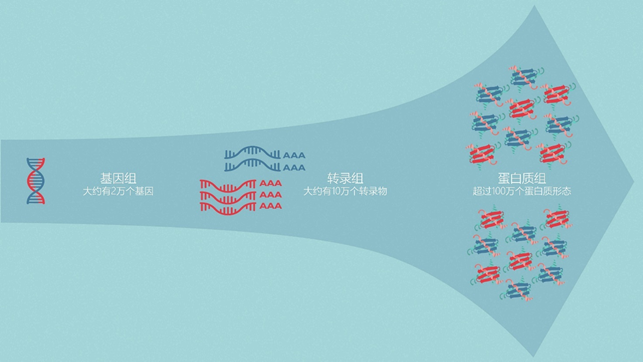


来源: 诊断科学
声明:本平台注明来源的稿件均为转载,仅用于分享,不代表平台立场,如涉及版权等问题,请尽快联系我们,我们第一时间更正,谢谢!


