



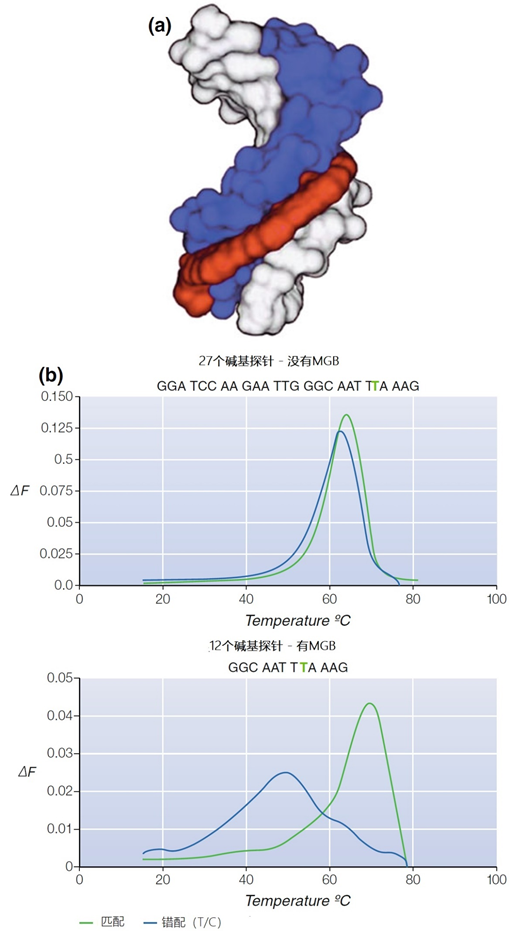
引物和探针的核苷酸序列至关重要,因为目标识别和后续步骤是通过杂交过程启动的。引物总是用化学方法合成的,长度在6个(随机六聚体)和大约100个碱基(用于PCR的复合引物)之间。 平均而言,引物的长度为19~25个核苷酸。引物可以在每个需要的位置上进行标记,例如用荧光素或生物素。另外,长度相当的探针,无论是否标记,都可以用化学方法合成。 除此之外,探针还可以通过分子生物学技术生产,如克隆或通过PCR扩增的方式。探针的大小可以有很大不同,而诊断(或研究)问题的定义决定了哪种探针最适合(图1)。 图1 | 杂交和检测杂交探针的命名法 一些表型的示意性概述: ➤ 寡头探针,在化学合成过程中进行标记,通常用于不同的独特探针的混合; ➤ 片段探针,通过重组DNA技术或PCR检测产生,通过酶技术进行末端标记; ➤ 总探针,通过重组DNA技术生产,通过随机引物或切口平移标记。 另外,选择的标签也是由问题决定的。稳定性,即引物或探针与目标之间的相互作用强度,可以通过应用5′-MGB(小沟结合)(图2)或加入锁定核酸(LNA)或蛋白核酸(PNA)来加强。也可以在PCR混合物中加入各种物质,以加强或削弱探针与目标的相互作用强度。 图2 | 小沟结合(MGB) a MGB是一种二氢环吡咯三肽,对DNA分子的小沟有特殊的亲和力。它增强了引物或探针的杂交能力。这样,只使用了普通数量的核苷酸的一半。 b中概述了传统探针和MGB探针的杂交和随后的融化过程。目标的大小是27bp,5′端第5位有错配(绿色野生型,蓝色T/C错配)。很明显,与传统探针相比,短的MGB探针具有更高的区分突变体和野生型的能力。 1、引物设计 引物在诊断学中主要用于扩增技术、cDNA合成、第二代测序和某些微阵列技术。只有PCR使用引物对组合,即两个可以在目标上下游退火的引物。这样,合成的核酸的大小就被确定了。可以使用以下引物的变体: ➤ 独特的引物使用独特的序列识别某个目标,在基因组或基因表达产物的其他位点上没有出现过。独特引物可用于PCR、测序和cDNA合成。独特引物可分为两类;完全匹配的引物和不匹配到一定程度的引物。后者可以专门设计,例如区分野生型和突变型基因组(错配鉴别)或能够检测一个(亚)物种或更高层次如家族的基因型中出现的一个或多个SNPs(错配耐受)。由于SNPs可以在复制过程中从新形成,特定的引物会失去其独特性。3′末端或附近的错配将在PCR中产生假阴性。 ➤ 非唯一引物可用于各种类型。重复引物粘附在多个位点上。突出的目标序列是微卫星,如(CACA)n-repeat、UTR或RE位点。Oligo-dT-引物用于真核生物的cDNA合成。这些引物与真核生物mRNA的3′-聚A-序列杂交。随机六聚体是用于cDNA合成、「缺口」翻译和微阵列的比较基因组杂交的通用引物。 ➤ 特殊引物,主要是独特引物的变体,在设计独特的引物时,世界范围内可用的数据库,主要是GenBank,和搜索工具(例如BLAST-基本局部比对搜索工具)是必不可少的资源。现在,越来越多的物种、基因型、甚至个体基因组的完整序列都是已知的。 对于开发新的诊断性PCR,诊断问题的定义是至关重要的: ➤ 需要识别哪个目标? ➤ 目标是一个基因、一个非编码片段、一个特定的突变甚至是一个易位? ➤ 是想识别一个病原体、一个抗性基因还是一组相关病毒的毒力因子? ➤ 是否需要确定一个肿瘤的表型或基因型或一个偏离的甲基化? 对于所有这些目标,重要的是至少要知道独特引物退火点的序列。随后,重要的是了解目标的性质,包括开放阅读框架(ORFs)的存在、组成(GC含量、保守片段的存在(有少量碱基变化)、二级结构和限制位点)、遗传(进化)稳定性,以及与突出序列的同源性,最好是BLAST控制。 随后,可以使用特定的(免费)(基于网络的)软件工具选择引物。更复杂的软件工具可在市场上买到,一般来说,不基于网络。 2、引物设计标准 独特的引物需要根据一些标准来设计,这些标准涉及引物的统一性、杂交条件下的线性、与目标物杂交时的稳定性、减少合成非特异性产物的变化等。许多这些标准被纳入独立的或在线的软件中;当引物不理想时给予惩罚,或对最佳引物给予高分。在此,以下要求是非常重要的: ➤ 引物内没有碱基配对(引物在杂交过程中是「开放卷曲」的)。 ➤ 没有GC-, GG-或CC-重复。 ➤ 应避免有超过3个相同碱基的重复(同聚物运行)。 ➤ 与3′端部的目标完美匹配。 ➤ 一个最佳的引物序列包含分布良好的核苷酸;即在位置和类型上都是如此。 ➤ 应防止正向引物和反向引物之间因互补碱基而杂交形成双联,特别是不应该有3′互补(图3a)。DNA聚合酶将产生引物二聚体,也被称为自体二聚体或交叉二聚体,从两个引物甚至小的双联区的3′-OH末端开始。即使没有3′-互补性,在反应物的不利比例下,如没有目标,也可能形成二聚体(图3b)。由于引物内其他核苷酸位置的互补性,引物之间的碱基配对也必须避免,因为这也可能导致引物二聚体的尺寸较小,熔化温度较低。虽然引物二聚体很容易被识别,但特异性扩增的效率会降低。 ➤ Tm值需要相似;最好在60℃左右,不低于56℃。 ➤ 引物的GC含量最好保持在45%以下,但应与完整的目标序列保持一致。 ➤ 优选的是,引物序列由分布广泛的核苷酸组成,并避免同聚物的运行。 ➤ 目标的引物退火位点必须是可接触的,最好不要位于稳定的发夹内。 ➤ 引物序列的理想长度为18-25个核苷酸,与所需的Tm和特异性有关。当使用LNA核苷酸或MGB时,要求的长度可能短至12个核苷酸。 ➤ 对于某些DNA,有可能设计出与两个连续外显子退火的引物,以防止与含有内含子序列的基因组DNA杂交。在这种策略中,基于内含子序列的引物设计是检测基因组目标的一种选择。然而,对假基因的鉴别是不可能的。由于许多细菌和线粒体DNA缺乏内含子,因此不可能设计出跨越内含子的引物。 ➤ 一对引物必须对其互补的目标序列有相当的亲和力(即Tm)。这将促进聚合过程中生长的新DNA链的稳定性。 ➤ 当测试样品含有目标DNA时,设计的引物需要产生正确长度的扩增物,而在非目标DNA样品中,不应该形成扩增物。 ➤ 探针检测中使用的引物对的Tm需要与探针的Tm一致,例如水解探针要低5-10℃(见第3.6.1节)。 ➤ 离子强度(Na+;Mg2+)、缓冲液(容量)、寡聚物的浓度以及如果有的话,PCR混合物中的DMSO必须作为新引物设计的参数。 如果在某些位置可能出现错配,可以将错配容忍度作为引物设计的标准。 图3 | 3′-互补性 a 一对引物的3′互补性导致两个引物的3′端相互杂交时合成引物二聚体。1987年在使用PCR检测镰状细胞贫血症的引物中首次描述了这种假象。 b 没有3个互补的引物二聚体可能是由于产品的不良比例造成的,如引物过多,特别是在没有目标的情况下(如目标的拷贝数很低或在阴性对照中)。引物二聚体(大小为30-50bp)可通过琼脂糖凝胶电泳(b;左)或熔解曲线分析(b;右)来观察(二聚体在70℃下熔解,扩增物在更高的温度下熔解)。 一般来说,引物被设计成与目标序列上的互补碱基完全杂交。第3.5节讨论了这一规则的例外情况。 3、探针设计标准 探针需要与一个互补的目标序列进行唯一的杂交。探针所针对的序列取决于(诊断)问题。例如,需要检测的是什么?为什么?检测的结果意味着什么?是基因组DNA、mRNA或rRNA还是具有诊断作用的miRNA之一?探针的大小和类型以及相应的杂交方法都存在很大的差异。 每种变体都是可能的;双链和单链的DNA或RNA探针,可以与DNA或RNA杂交。这意味着DNA:DNA、DNA:RNA或RNA。RNA的组合可能发生。根据GC比率的不同,形成稳定的杂交体所需的最小长度是13-20个核苷酸。理论上,没有设定最大长度;有时,大于400个碱基的探针会被分解(图1)。 一般来说,反应条件要根据每个特定的探针-目标组合以及反应发生的基质来调整。需要在大小、核苷酸序列和目标物浓度,以及产生足够信号所需的探针量之间取得平衡。 探针的杂交能力描述了探针与目标结合的能量。简而言之,两个互补的(多/寡)核苷酸链内和之间的所有非共价相互作用的总和表示为ΔG(吉布斯自由能),单位为kcal/mol。这个数值越负,探针与目标的结合就越强。 正值表示需要添加能量,不会自发发生反应。促进探针杂交能力的因素是探针的G/C含量和大小。富含A/T的寡头探针将更容易从其目标上解离。最好是探针的G/C含量与目标序列周围的区域相似,特别是在基于探针的PCR中。 如前所述,对探针的最大长度没有限制。在分子诊断中,探针用于鉴定原始或纯化的样品、细胞或组织样品中的目标DNA,无论是否在扩增目标(s)之后。用于PCR的探针的正常长度约为15-30bp。 如果在PCR反应中使用,这些探针通过杂交确认扩增物的存在。特别是,实时PCR的引入导致了水解探针、「双」探针和分子信标的大规模使用,成为确认扩增物身份的重要工具。这尤其适用于微生物学和突变检测研究。 适用于筛选DNA数据库和染色体分析的探针可能有数千个核苷酸长。几乎在所有情况下,碱基序列都需要知道。然而,「全染色体」杂交、SNP阵列或比较染色体杂交(CGH)则不需要。当序列已知时,探针的设计与引物的设计是相同的。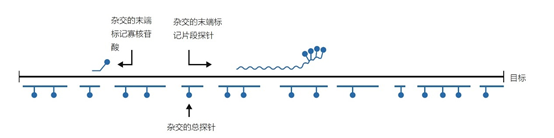

来源: 诊断科学
声明:本平台注明来源的稿件均为转载,仅用于分享,不代表平台立场,如涉及版权等问题,请尽快联系我们,我们第一时间更正,谢谢!


