



1
伴随诊断的验证
当CDx的临界值被选定后,就可以最终完成分析验证,在此,必须证明该检测方法准确可靠地测量有关的生物标志物。当一种检测方法要在临床试验中前瞻性地选择患者时,重要的是它要证明有足够的分析稳定性,特别是在选定的临界值附近。
一般来说,进行分析验证研究是为了评估CDx的任何关键性能特征。通常情况下,分析验证必须在检测方法用于关键的临床试验之前完成,该试验将支持与药物诊断组合有关的声明。
最终的检测方法必须在符合GMP的条件下生产,因此,随后对检测方法的分析验证将提供客观的证据,证明根据预期的用途已经达到了设计规格。
最终的检测配置和设计是通过生成准确度、灵敏度和特异度、稳健性以及一些不同的精度和重现性研究(如检测内和检测间变异性、批次间变异性和仪器间变异性)的数据来验证的。
作为分析验证的最后一部分,通常要进行外部重现性研究,以记录日常、读取器间和实验室间的重现性。这个外部研究的地点是在代表CDx检测的终端用户的临床实验室中选择的。
与分析验证相关的不同活动对于确保CDx测定满足其规格要求至关重要;因此,它可以以安全和有效的方式部署在临床环境中。
CDx测定也可以在单个实验室内开发使用,这样的测定被称为实验室开发的测试(LDT)。目前,这种类型的检测方法有两种,即BRACAnalysis CDx(Myriad Genetic)和FoundationFocus CDxBRCA Assay(Foundation Medicine),与两种不同的PARP抑制剂奥拉帕利(Lynparza,AstraZeneca)和鲁卡帕利(Rubraca,Clovis Oncology)相连,已经获得美国FDA批准为CDx。
然而,与其他CDx相比,LDT只能在开发该检测方法的实验室内使用,而且必须在CDx检测的使用说明中指明这种指定实验室的名称。
2
临床试验方案设计
纵观成功完成的药物-诊断试剂联合开发模式项目的数量,在涉及CDx测定的临床验证时有一个共同点,那就是富集试验设计。
在这里,研究人群是根据CDx测定的测试来选择的,而且只有生物标志物「阳性」状态的患者才会被纳入试验中。
然而,只有在有明确的证据表明生物标志物阳性状态与研究药物的疗效之间有密切的关系时,才可以使用富集设计,例如,从以前进行的临床前和临床研究中。如图1 A和B所示,这种试验设计的随机和非随机版本都被采用。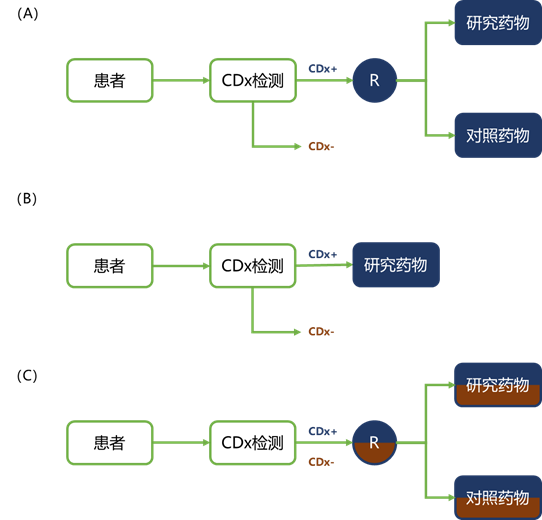
图1 | 临床试验设计。(A) 随机富集的设计。只有测试阳性的患者被随机分配到研究药物或比较药物。(B) 非随机设计。只有检测阳性的患者接受研究药物的治疗。(C) 随机化全员研究。所有患者都被随机分配到研究药物或比较药物,与CDx检测结果无关。CDx,同伴诊断法;CDx-,测试阴性;CDx+,测试阳性;R,随机。
通过CDx检测对患者进行预选,减少了群体内的变异性,增加了可能有反应的患者的比例,从而提高了研究力量。
如我们在前面的系列文章中所描述的,当曲妥珠单抗及其CDx检测被批准用于治疗HER2阳性的转移性乳腺癌妇女时,就采用了随机富集设计。
类似的试验设计随后被用于其他癌症靶向药的临床验证和批准,如vemurafenib(Zelboraf,Roche/Genentech)和pertuzumab(Perjeta,Roche/Genentech),以及它们各自的CDx检测。
随着一些CDx检测的预测能力的提高,一种更简单的富集设计被引入,即单臂富集研究。
近年来,这种简单的试验设计已被用于临床验证和获得几个药物诊断组合的监管,如克唑替尼(Xalkori,辉瑞)/Vysis ALK Break Apart FISH Probe Kit(雅培分子),以及鲁卡帕里(Rubraca,Clovis Oncology)/FoundationFocus CDxBRCA测试(Foundation Medicine)。
尽管富集试验设计往往能够在相对较少的患者中提供关于药物疗效的结论性答案,但它也有缺点,其中之一是它没有提供关于CDx检测阴性患者的药物疗效信息,因此,诊断指标的计算也受到限制。事实上,富集试验设计只允许计算PPV。
如前所述,只有在有数据支持生物标志物状态和药物疗效之间有密切关系的情况下,才应使用富集试验设计。如果这类证据很弱或不存在,则应采用分层或全部参与者的试验设计,如图1 C和图2所示。
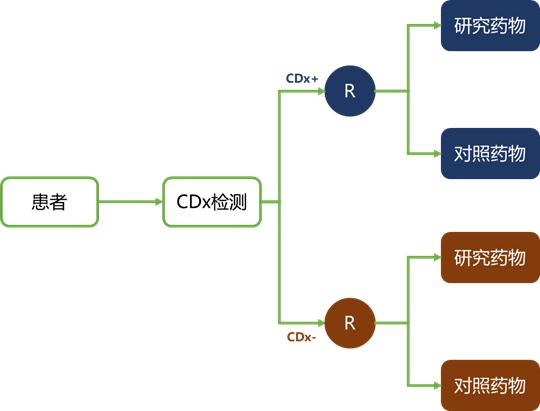
图2 | 分层试验设计。所有患者根据CDx检测结果进行分层,随后随机分配到研究药物或对照药物。CDx,伴随诊断;CDx-,测试阴性;CDx+,测试阳性;R,随机。
所有参与者的设计没有将生物标志物的阳性状态作为纳入标准的一部分,测试阳性和测试阴性的患者都可以进入研究,随后随机分配到研究药物或对照药物。
大多数情况下,这种试验设计用于早期阶段的研究,在这些研究中,药物疗效与生物标志物状态之间的联系是不确定的。使用全员试验设计既有优点也有缺点。
从正面看,这种设计可以计算不同的诊断指标,如灵敏度、特异度、PPV和NPV。
然而,也有一个明显的缺点。如果检测阳性患者的发病率很低,就需要很大的样本量来得出CDx检测的预测潜力的结论。
以类似的方式,分层试验设计将生物标志物阳性和阴性的患者都纳入到不同的研究臂中,然后他们被随机分配到研究药物或对照药物中。
这种设计或多或少类似于进行两个独立的随机试验,一个在检测阳性患者中,一个在检测阴性患者中;但是,在这里,它们是在同样的后勤体系下进行。
与所有参与者的设计相比,这里更容易管理测试阳性和阴性患者群体中可能存在的患病率差异,因为他们属于研究的不同部分。此外,对于所有参与者的研究,有可能计算出CDx检测的不同诊断指标。
对于任何CDx来说,关于不同诊断指标的信息对于评估检测的临床有效性是很重要的。任何应用CDx检测的医护人员都应该知道其灵敏度和特异度,以确定对有关药物的反应者和非反应者。
计算这些不同的诊断指标通常需要获得二元的结果指标,正如在肿瘤学临床试验中所知道的那样,根据RECIST标准的客观反应可以将患者分为反应者和非反应者。
然而,由于这些试验大多依赖于事件终点的时间,如总生存期或无病生存期,最近有人提出了一种使用这些结果变量计算不同诊断指标的方法。
希望这样的建议能使更多的CDx检测具有可获得的灵敏度、特异度、PPV和NPV的数据。
事实上,关于PD-L1 IHC 22C3 pharmDx检测的灵敏度和特异度数据已经公布,这与选择NSCLC患者接受pembrolizumab治疗有关,是一个值得借鉴的例子。
3
临床验证
临床验证的目的是证明CDx检测能够检测患者样本中感兴趣的生物标志物,并确定有望从研究性药物中获益的患者群体。
临床验证最好包括对临床诊断指标的评估,如灵敏度、特异度、阳性预测值(PPV)和阴性预测值(NPV)。这种评估是否可能取决于所选择的试验设计,这一点将在后面讨论。
通常情况下,在分析验证结束之前,CDx检测的临床验证不应启动。重要的是,关键的临床试验要用「最终版本」的检测方法进行,因为分析性能会影响患者的选择。
就药物开发而言,为了达到最佳的治疗效果,让合适的患者接受研究性药物是至关重要的,对于患者来说,知道有公平的机会从新疗法中获益是很重要的。
然而,由于药物和CDx开发的一致性和时间上的挑战,有时很想用原型测定法开始关键性的临床试验,然后在试验后期用分析验证的版本取代它。不幸的是,由于几个原因,这种策略是不值得推荐的。
首先,根据未经验证的检测方法选择患者,有可能出现假阳性和假阴性的检测结果,这可能导致错误的分类。
第二,它使临床研究的结果难以解释,因为它把患者分成两个亚群;一个是用原型检测法选择的人群,另一个是用最终分析验证的版本选择的人群。
如果在关键的临床试验中使用了不同版本的检测方法,就需要进行后续的衔接研究,这对资源和时间的要求都很高,而且会给药物-诊断试剂联合开发模式项目增加额外的风险。
CDx检测的临床验证通常是在关键临床试验中证明研究药物的安全性和有效性的同时进行的,在此,重要的是要记住「一个规则」,即只使用一个检测版本,即最终分析验证的版本,此外,只使用一个测试点,以减少可能的实验室间差异。
好了,以上就是今天分享的全部内容,如果大家有什么疑问,欢迎在后台给我们留言,或者加入我们一起讨论,若觉得文章不错,也请关注我们,以免错过后续更新~
来源:诊断科学
声明:本平台注明来源的稿件均为转载,仅用于分享,不代表平台立场,如涉及版权等问题,请尽快联系我们,我们第一时间更正,谢谢!


