



在上一篇文章当中,我们介绍了核酸的分离和纯化,今天我们将介绍PCR的干扰因素。
由于PCR的灵敏度非常高,污染被认为是影响PCR结果的最重要因素之一,会产生假阳性结果。
同样关键的是导致假阴性结果的各种来源。如果PCR混合物或扩增反应本身的一个或多个基本部分受到抑制或干扰,诊断性检测就会受到阻碍。这可能导致效率降低,甚至出现假阴性结果。
除了抑制之外,在样品制备之前,由于运输和/或储存条件,目标核酸的完整性可能会发生损失。特别是,高温或保存不充分可能导致细胞和核酸的损坏。
细胞和组织固定和石蜡包埋是众所周知的造成DNA破碎的原因,也是一个持久的问题,我们也将会在后面的文章当中专门讨论。在这些情况下,即使是最佳的分离和纯化也无济于事。
在分子诊断中经常发生的另一个常见的问题是,与苯酚-氯仿提取相比,目标核酸的释放不够理想。在极端情况下,这可能与假阴性有关。通过煮沸裂解或酶解细胞碎片可以节省很多时间,但这种方法往往由于核酸释放不足而导致PCR灵敏度低。
1、扩增过程中对聚合酶活性的抑制
一般来说,抑制其实是一个框,我们将所有导致PCR结果不理想的因素都装在这个框里。在严格的生物化学意义上,抑制仅限于酶的活性,即通过与DNA聚合酶的活性位点或其辅助因子(如Taq DNA聚合酶的Mg2+)的相互作用,减少或阻止底物-产品的转化。
样品中的成分或含有试剂的各种缓冲液和提取液可以直接抑制酶或捕获其辅助因子(如EDTA),从而使聚合酶失活,反过来导致PCR结果下降或假阴性。
然而,反应成分和含有目标的核酸之间的许多相互作用也被指定为「PCR抑制剂」。一旦细胞的完整性被隔离破坏,核酸被释放,样品与其周围的溶液和固相之间就会发生相互作用。
例如,「清道夫」可以通过非共价作用结合单链或双链DNA,并通过减少最终到达PCR反应的目标数量来干扰分离和纯化。
在实践中,这导致了与酶抑制本身相同的读出结果。
试剂管、膜或珠子的截留也会造成同样的结果;目标物的回收率低于100%是一个众所周知的现象,这取决于所应用的分离和纯化方法。
尽管提取液的目的是快速破坏核酸酶,但休息时的活动可能会破坏目标物,如在储存和/或运输过程中会发生。甚至插层染料(如SYBR® Green I)通过推测增加双链构型的稳定性对PCR反应的干扰也被命名为PCR抑制。
已经有大量的PCR抑制剂被发现,但这些化合物的身份和作用并不总是很清楚。
一般来说,PCR抑制剂存在于大多数体液和用于临床诊断测试的试剂(尿液中的尿素、血液中的血红蛋白和肝素)、膳食补充剂(有机成分、糖原、脂肪、Ca2+-离子)和环境中的成分(酚类、重金属),具体可见表1。
表1|来自不同来源的PCR抑制物质的知名例子
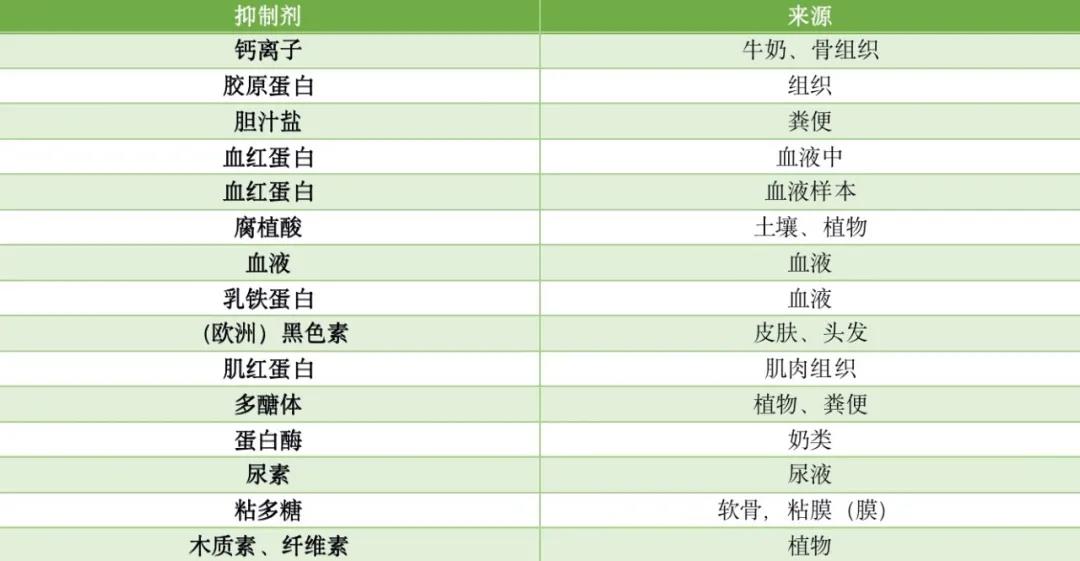
更普遍存在的PCR抑制剂可以在细菌和真核细胞、非目标DNA、组织基质的DNA结合大分子和实验室设备(如手套和塑料)中找到。
在提取过程中或提取后对核酸进行净化是去除PCR抑制剂的首选方法。如今,各种自动化提取设备可以取代许多人工方案,但目标的100%回收和/或纯化从未达到。
潜在的抑制剂可能仍然存在于纯化的核酸中,或者已经发生了作用。
存在不同的策略来减少抑制剂的影响。选择合适的聚合酶可以对抑制剂的活性产生重大影响。其他被证实的减少PCR抑制的方法是增加聚合酶浓度或应用添加剂,如BSA。
PCR反应的抑制可以通过使用内部过程控制(IPC)来证明,我们将会在后续的系列文章当中专门讨论这个问题
必须注意通过彻底的清洗步骤从核酸分离物中去除提取试剂盒中的所有试剂和其他溶液,如乙醇、EDTA、CETAB、LiCl、GuSCN、SDS、异丙醇和苯酚。根据它们的浓度,它们可能激活或抑制PCR。
2、石蜡包埋样品中核酸的完整性
在病理学、组织学和细胞生物学中,大多数组织样本都是通过化学方法固定的,以保护它们不被自溶过程和腐烂所破坏。
此外,保存可以稳定结构,以确保形态学细节。这意味着结构成分,如蛋白质、核酸-蛋白质复合物和脂质-蛋白质复合物,被制成不溶性。
在病理学上,福尔马林是最常用的固定剂。
固定后的样本随后被嵌入石蜡中,用于制备显微镜下的切片和存档。
石蜡包埋的切片也可作为核酸提取和后续分子诊断的样本。当然,这意味着在进行DNA/RNA分离之前,需要去除石蜡并对切片进行补水。
然后,通过福尔马林固定的石蜡包埋(FFPE)组织的特殊技术裂解组织,该技术还可以通过蛋白酶破坏福尔马林-蛋白质桥,特别是核酸/组蛋白复合物。
离心后,上清液中存在非纯化的核酸。虽然这种粗分离物可用于许多分子分析,但在大多数情况下,它将被纯化,以去除蛋白质和膜的残留物,例如,基于Boom技术的自动程序。纯化的核酸确保了更高的分析质量并增加了耐久性。
福尔马林固定的一个很大的缺点是核酸的碎片化,这阻碍了各种分子诊断技术的应用,如PCR和测序。
破碎的程度与固定的时间和随之而来的对机械破坏的抵抗力有关,特别是在56℃的石蜡浸润期间的gDNA。
建议至少在缓冲福尔马林中固定<3 mm的组织切片24小时。对可能发生在多个外显子的基因突变的分析,需要分成多个小的PCR产物,用序列分析法进行分析。
例如,肿瘤抑制基因p53由6个外显子组成,使用8个PCR产物进行分析,在正向和反向方向进行测序。
在分析之前,通过使用Ladder PCR测试DNA质量。这是一种针对参考基因(在所有细胞中表达相同的基因)的多重PCR,其PCR产物大小不断增加,以确定可从DNA样品中扩增的最大PCR产物。
对于在固定位置(热点)有突变的基因,其他突变分析方法更适合。这些方法包括定量PCR(qPCR)和高分辨率熔解分析(HRMA)分析,这些方法对高达150个核苷酸的PCR产物起作用。
注:Ladder PCR是一种扩增不同大小(长度为碱基对)的基因组或mRNA目标的方法,以评估单个FFPE分离物作为特定目标模型的质量。由此,可以选择RT-(q)PCR、FISH或CISH的方法。
RNA越来越多地在分子病理学中发挥重要作用。除了固定的问题,在使用RNA时,被RNases快速降解是另一个重要问题。
为了避免这个问题,RNA在纯化后立即被转化为更稳定的cDNA。这样,根据RNA的原始质量,可以扩增出高达200bp的PCR产物。因此,来自固定样品的RNA不太适合于序列分析。
在淋巴瘤和肉瘤的分子诊断中,检测易位的一个重要目标是肿瘤样本的mRNA。
转位源于位于不同染色体上的两个基因的融合,可导致一个新的融合基因,是某一类型肿瘤的特征。RT-(q)PCR可以检测在这种细胞中产生的mRNA,但为此必须要有完整的mRNA,所以首选冷冻切片。
如果在石蜡切片中不能检测到易位,使用肿瘤细胞染色体DNA上的探针进行荧光原位杂交(FISH)是处理严重碎片化的mRNA的首选方法。
好了,以上就是今天分享的全部内容,如果大家有什么疑问,欢迎在后台给我们留言,或者加入我们一起讨论,若觉得文章不错,也请关注我们,以免错过后续更新~
来源:诊断科学
声明:本平台注明来源的稿件均为转载,仅用于分享,不代表平台立场,如涉及版权等问题,请尽快联系我们,我们第一时间更正,谢谢!


