



体外诊断试剂注册与备案管理办法的基本情况
在本文中,将会给大家介绍一下体外诊断试剂注册与备案管理办法的基本的框架。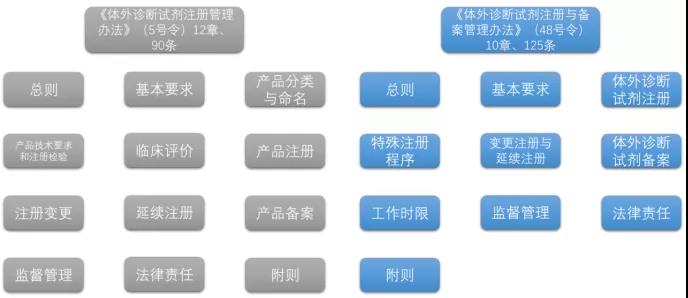
图1|新旧体外诊断试剂注册管理办法框架对比
《体外诊断试剂注册与备案管理办法》(48号令)10章125条,而《体外诊断试剂注册管理办法》(5号令)是12章90条,与2014版的办法相比,增加了三十五条,增加的幅度接近40%。
其中框架上主要变化包括删除了原办法中产品分类与命名这样一个章节,它将会作为一份单独的规范性文件,另行出台。
同时将产品技术要求和注册检验、临床评价和产品注册这三个章节合并为体外诊断试剂注册一章,将产品注册和注册变更章节合并为变更注册与延续注册一章。这里需要注意的是,在体外诊断试剂注册一章中,增加了产品研制这样一个章节。
这是借鉴和参考医疗器械注册与备案管理办法的章节设置,同时也增设了特殊注册程序一章,规定了产品创新产品、优先产品和应急产品的特殊注册程序,也增设了工作时限一章。
体外诊断试剂办法与医疗器械办法相比,相同的条款数是104条,大约占了5/6,而体外诊断试剂特殊的条款有21条,大约占1/6的这样一个比例。
这些不同的条款主要集中在以下方面,包括:
定义和范围;
产品技术要求;
体外诊断试剂非临床研究的内容;
体外诊断试剂不同包装规格检验的要求;
检验报告的要求;
使用国家标准品的情形和要求;
体外诊断试剂临床评价要求;
审批和注册证发放;
体外诊断试剂变更要求;
不属于变更的情形;
不予延续的情形;
体外诊断试剂产品命名要求;
注册单元划分要求;
组合部件销售;
临床机构自研试剂。
接下来,我们将对体外诊断试剂注册与备案管理办法当中一些重点的内容进行介绍。
体外诊断试剂注册与备案管理办法重点内容介绍
2.1、体外诊断试剂的分类
在5号令当中,有四条内容是涉及到体外诊断试剂产品分类的,包括第十七条,IVD产品的分类原则,规定了体外诊断试剂产品分类的一个基本原则。
第十八条,第二类产品按第三类产品注册的情形,是第二类体外诊断试剂当中哪些情形需要按照第三类体外诊断试剂进行管理的。
第十九条,校准品、质控品的分类原则和第二十条,分类目录制定和调整。
这四条内容,在48号令当中,都进行了删除,合并为一条,就是体外诊断试剂注册、备案工作应当遵循体外诊断试剂分类规则和分类目的有关要求。
那么,为什么会做这种这样的一个删除呢?
主要依据主要还是考虑到现行的体外诊断试剂行业发展的情况,以及NMPA近年来对体外诊断试剂管理类别调整的现状,让法规能够更好的适应产业发展的情况,可以更快更便捷的根据产业发展和监管的需要,对产品的管理类别及时的进行调整。
所以这一部分内容呢,被单独拿了出来,不在配套规章中进行相关产品的具体表述。
这样更便于今后对体外诊断试剂产品分类工作进行动态的调整和进一步的细化完善。
出于这种方面的考虑,在办法里做了这样的修订,NMPA为了进一步做好体外诊断试剂产品相应分类的工作,配合试剂办法的出台,在今年5月19日发布了《体外诊断试剂分类规则(征求意见稿)》,向全社会公开征求意见。
新的体外诊断试剂分类规则,参考了IMDRF最新成果文件,如对诊断和治疗的影响、个人和/或公共健康的影响,使用者的专业知识等等。
也结合了过去NMPA对一些产品的类别调整的成果,例如肿瘤治疗过程监测的一些产品,在2020年的管理类别进行的调整,以及免疫组化类、流式细胞仪及配套使用的产品、过敏原相应的产品在2017年进行的调整。
这些最新的调整成果,也都纳入了新的分类规则当中。
同时还结合了一些日常分类界定的经验,包括反应通用试剂、伴随试剂等等。
2.2、产品研制
借鉴医疗器械注册办法的内容,在体外诊断试剂注册办法中增加了产品研制一章。这一章分为四节,包括编制技术要求、编制说明书和标签、非临床研究和注册检验。
2.2.1、编制技术要求
在编制技术要求这一节当中,进一步明确了申请人、备案人应当编制申请注册或者进行备案体外诊断试剂产品的技术要求。
产品技术要求,应该包括可以对体外诊断试剂成品进行客观判定的功能性、安全性指标和检验方法。
在体外诊断试剂产品技术要求中,有一个的特殊要求,就是第三类体外诊断产品,需要提供技术要求附录。
在5号令中,要求附录中,需要明确主要原材料、半成品以及生产工艺要求。而在48号令中呢,为了更好的体现技术要求是对产品成品质量进行控制,而不是对生产过程质量进行控制的作用,在附录当中删除了对半成品的相应的要求。
2.2.2、非临床研究
在48号令当中,结合体外诊断试剂产品的特点,明确了体外诊断试剂非临床研究的相关的内容。
首先,是明确了它的定义,就是在实验室条件下对体外诊断试剂进行的实验或者评价。
接下来进一步明确了非临床研究的主要内容,这部分内容和5号令是一致的,包括原材料的选择及制备、产品生产工艺、产品分析性能、阳性判断值及参考区间和产品稳定性等方面。
同时也明确了非临床研究,是应该结合产品的预期用途和技术特征来开展的,也就是说要结合产品的具体情况来开展非临床研究。
最后呢,明确了申请注册或进行备案,应提交非临床研究证据的要求。这个是结合体外诊断试剂产品的特点,对非临床研究研究内容进行一个细化。
2.2.3 注册检验
注册检验的内容和器械办法是一样的,为了落实新条例当中关于注册检验相关改革的要求,在48号令中,也明确了注册人提交的检验报告可以是申请人、备案人的自检报告。也可以是委托有资质的医疗器械检验机构出具的检验报告。
这个改革力度是非常大的,尤其是对于体外诊断试剂产品而言,它带来的利好要大于医疗器械产品。
因为对于医疗器械产品而言,很多产品的检验工作是企业自己是无法完成的,例如EMC、电器安全等等。
但是对于诊断试剂产品而言,因为产品的特殊性,可能多数的企业都具备基本的检测硬条件,所以很多体外诊断试剂厂家,就认为是不是我用自己的实验室,随便做一个全项检验就可以提交产品的注册检验报告了呢?
这答案可能是否定的,对于开展自检工作的注册申请人或者是备案人,一定要落实企业的主体责任,要具备相应的质量体系和相应的检验能力。
在这种条件和能力均具备的情况下,才能开展相应的自检的工作,如果不具备这种条件和能力还是应该委托有资质的医疗器械检验机构出具相应的检验报告。
第二个方面,是关于产品批次要求的,在5号令中也有相应的要求,就是要求三个批次的产品的检验报告。
这个是由体外诊断试剂的特点决定的,主要是由于体外诊断试剂产品生产批次之间的批间差有的时候会很大,所以设定了相应的要求。
但是在这次修订过程中,删除了连续三个批次的要求,这也是出于一个科学的考虑,试剂上连续三个批次,实际上并不能充分的反映产品的批间差,所以实际上,之前的规定呢,对于产品性能考核来说,反而更宽松,所以在48号令中,不再要求连续三批。
国家标准品、参考品也是一个重点关注的内容,一方面是由于国家标准品和参考品更新有些滞后,不适用与一些新产品,所以呢,有很多厂家希望不再对标准品、参考品的来历进行限定,扩大这个要求范围。
但是呢,国家标准品、参考品对于体外诊断试剂的质量评价和质量控制,都起着非常重要的作用,是重要的标尺,因此,在48号令当中,保留了这一条款,但是在文字表述上也做了修订,增加了「有适用的」这样的描述性定语。
这是考虑到在试剂使用标准品检验的过程中,由于方法学的不同,或者一些新的方法学的出现造成了相应的标准品的不适用于新出现的产品,使得新出现的产品无法通过标准品的情形,因此呢,加入了这样的限定。
另外将参考品进行了删除,这主要是由于参考品的概念被并入了标准品中,所以国家标准品是包含国家标准品和以及相应的参考品的。
作者:认真的刘博
责编:安悦
信息来源:诊断科学
声明:本平台注明来源的稿件均为转载,仅用于分享,不代表平台立场,如涉及版权等问题,请尽快联系我们,我们第一时间更正,谢谢!


